Research
All organisms must maintain genome integrity by reversing DNA damage caused by cellular metabolism, pathogens, radiation, or chemicals. Breaks in both DNA strands – Double Strand Breaks – can severely damage or kill cells. Cells respond to these lesions by engaging DNA repair pathways. These pathways are distinct, require specialized repair factors, and produce characteristic repair outcomes. How cells “choose” the right DNA repair pathway for each DSB remains an unanswered and fascinating question. Beyond simply reversing damage, the introduction and repair of DSBs is also crucial for cellular biology. Diverse cellular processes from DNA replication to meiosis proceed through DSB intermediates. Our goal is to understand the regulation of DSB repair, which we expect to not only reveal fundamental biological insights into how cells fix damage, but also translate to improved gene editing outcomes or cancer treatments. Research projects in the lab are oriented around three questions: 1) What role do Fanconi Anemia proteins play in DSB repair; 2) Does DSB repair vary in a tissue-specific manner; and 3) What role does DNA replication play in DSB repair pathway choice?
What role do Fanconi Anemia proteins play in DSB repair?
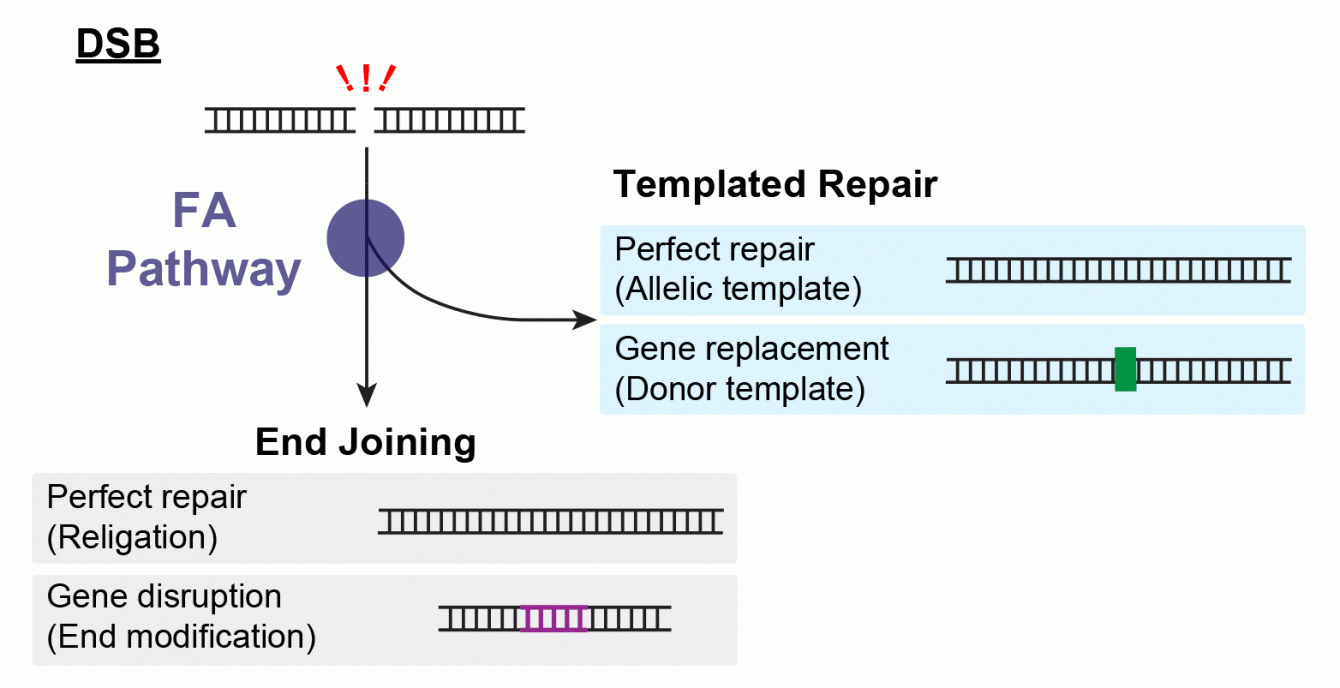
DNA Double Strand Break repair pathways work by end-joining, in which both ends of the break are stuck back together, or templated repair, in which information from a template molecule is used to copy sequence spanning the break site. The Fanconi Anemia pathway is required for templated repair and components of this pathway localize to DSBs during DNA repair. Together, these results suggest that Fanconi Anemia proteins play key roles in the “decision” between End Joining and Templated Repair. Ongoing work in the lab will uncover the precise role played by a key Fanconi Anemia protein, FANCD2, during DSB repair. We are investigating this topic using molecular biology, genetic, and biochemical approaches.
Does DNA repair vary in a tissue specific manner?
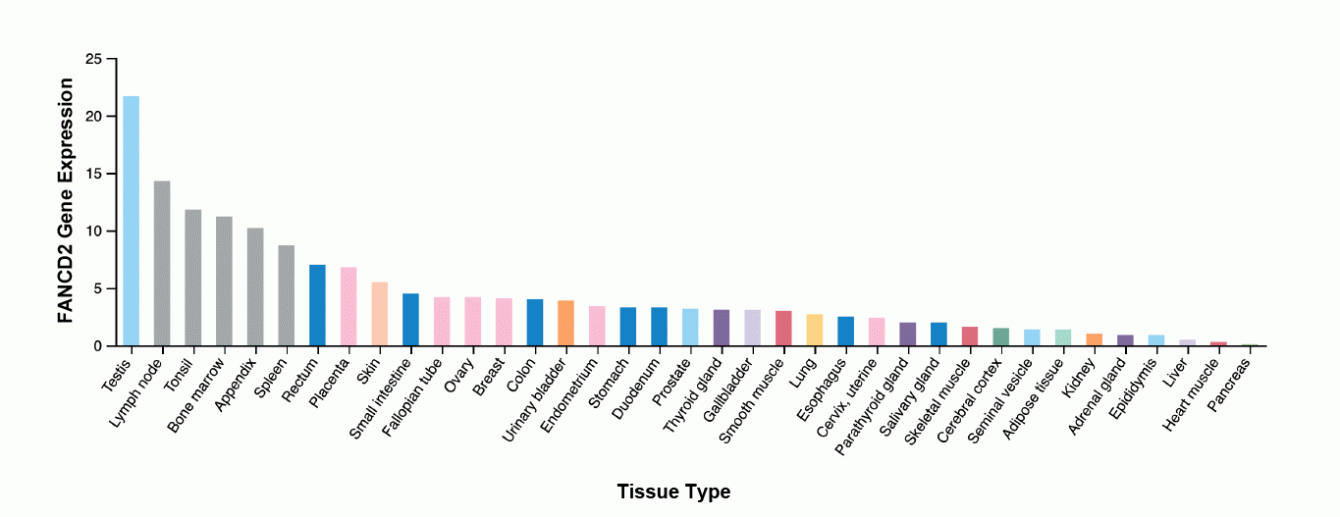
All cells must repair their genomes. However, cells in different tissues express different levels of DNA repair proteins. For example, FANCD2, which is important for Double Strand Break repair, is up-regulated in some tissues (e.g. bone marrow) and absent in others. This observation raises intriguing biological questions: 1) Do cells expressing little to no FANCD2 implement different DSB repair strategies; 2) Why do cells upregulate DNA repair factors in certain tissues and not in others; 3) Is upregulation of DSB repair proteins driven by tissue-specific stress and, if so, what are the stresses; 4) Do expression profiles (and by extension DNA repair strategies) change in a given tissue as the product of stress, metabolic change, or disease? We are using bioinformatic, genetic, and molecular biology approaches to answer these questions.
What role does DNA replication play in DSB repair?
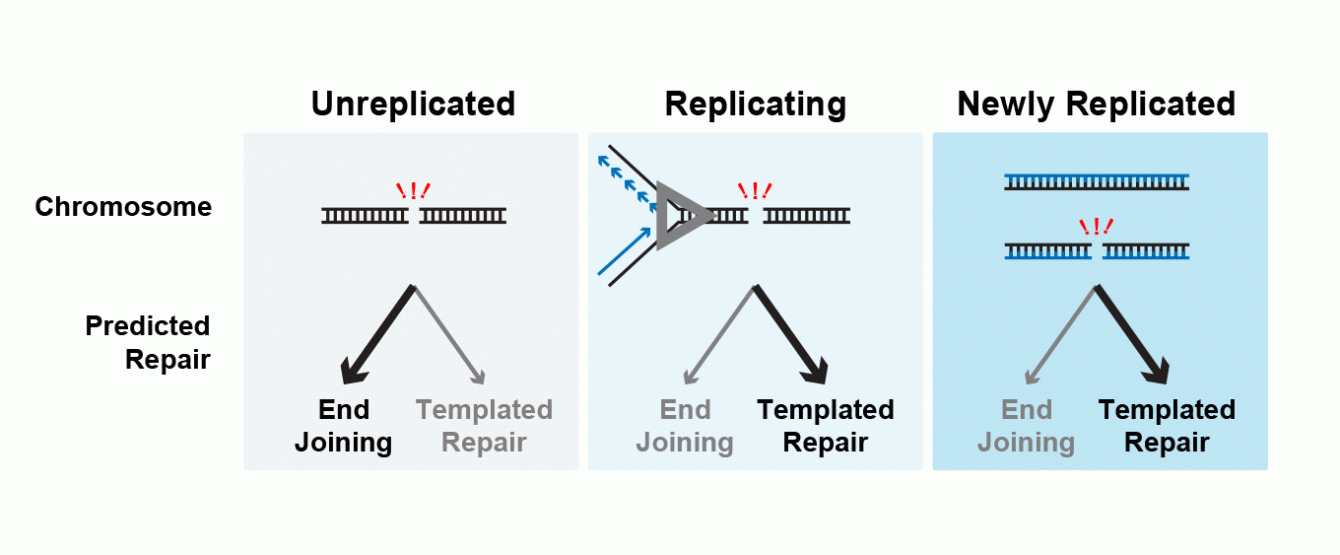
DNA damage and repair occur on active chromosomes that are also undergoing transcription, replication, and epigenetic alteration. Cells must therefore coordinate between DNA repair and other metabolic processes. We want to understand how one of these processes, DNA replication, influences Double Strand Break repair. These two cellular activities are known to be tightly linked: the abundance of DNA repair factors increases during replication to compensate for the additional DNA damage caused by this process, and the activity of many DNA repair proteins is altered by cell cycle signaling. These observations lead to a model that DSBs are repaired by one type of DNA repair (End Joining) prior to DNA replication and another type of DNA repair (Templated Repair) after DNA replication. The simplicity of this model obscures that DNA replication is progressive and replicating cells proceed from mostly un-replicated to mostly replicated. Therefore, DSBs in a replicating cell may be detected and repaired while the chromosome is unreplicated, as the chromosome is being replicated, or after the chromosome has been replicated. Current measurement techniques average these separate categories, which may obscure interesting biology. We have developed a molecular biology/genomics approach that allows us to measure DNA repair outcomes at each individual DSB and simultaneously determine if it was repaired while the chromosome was unreplicated, partially replicated, or fully replicated. This allows us to understand if DNA repair preferences are altered by replication itself or by another form of regulation, for example cell cycle signaling.